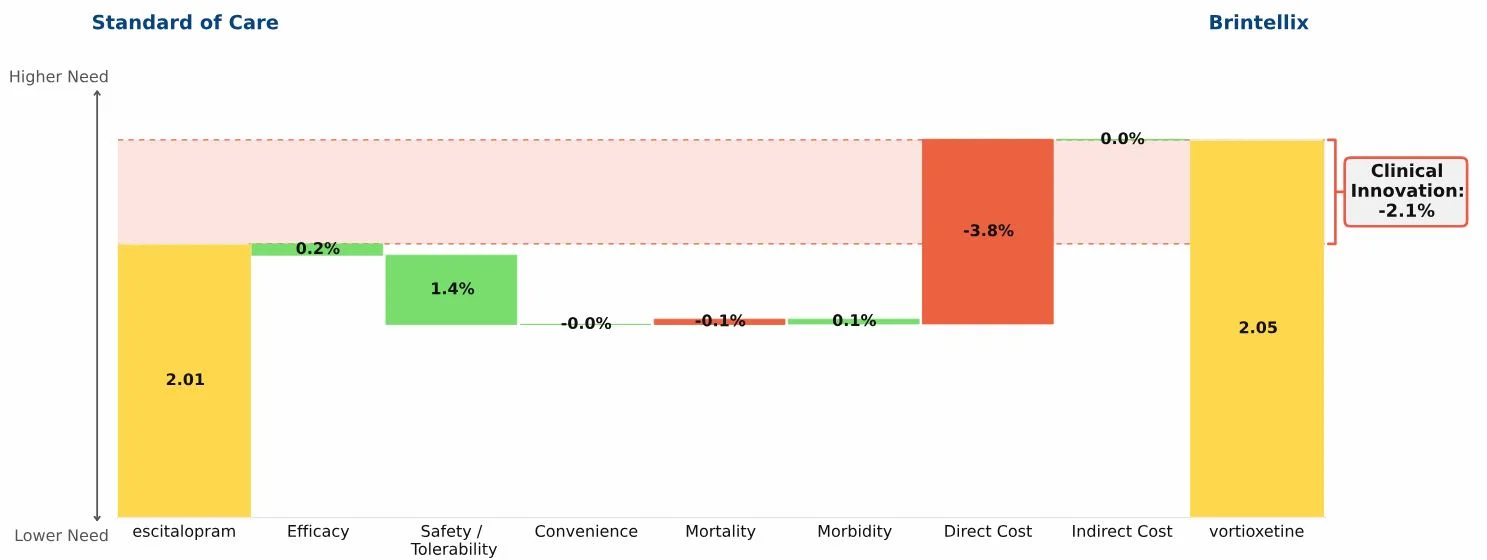
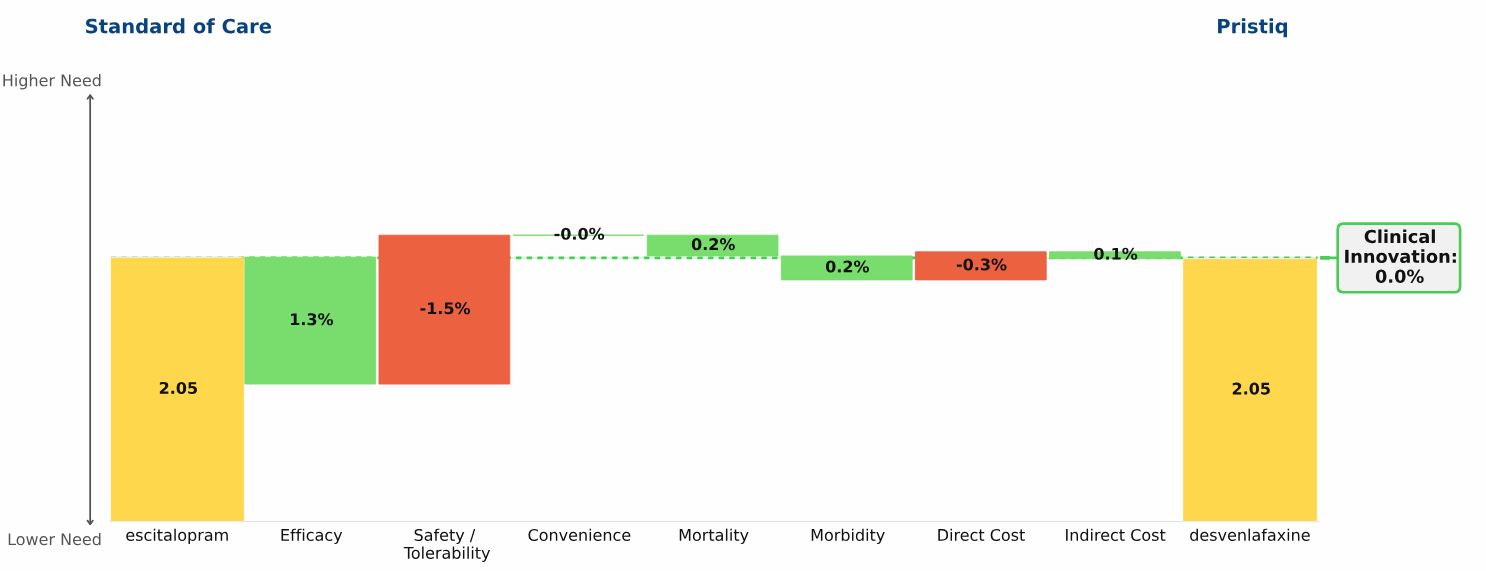
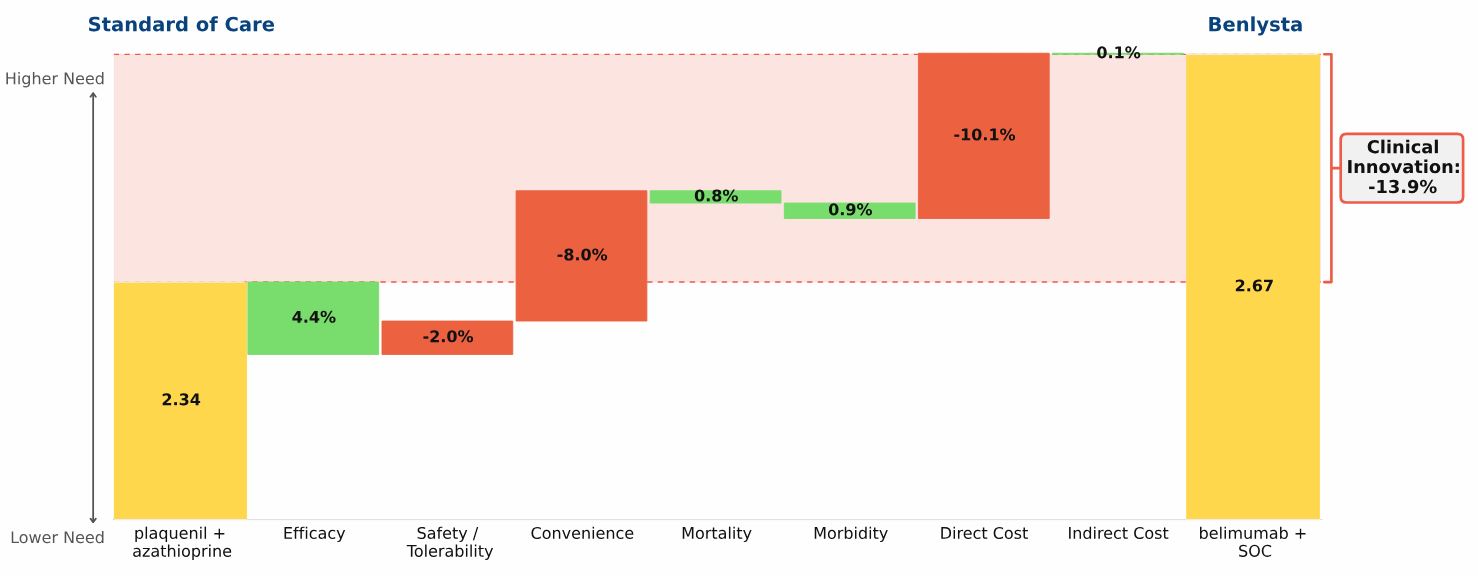
Conclusion: Three new oral drugs launched in recent years to treat multiple sclerosis, Gilenya, Aubagio, and Tecfidera, each offer clinical advantages over the beta interferons; Tecfidera has the greatest advantage, followed by Gilenya, and Aubagio has the smallest clinical advantage. Commercial performance has closely tracked these differences in Clinical Innovation.
Several new drugs have entered the multiple sclerosis market over the past few years. Gilenya (fingolimod) has strong Clinical Innovation at 9.3%, when compared with Rebif (interferon beta-1a). The “Drivers of Improvement” graphic below summarizes the results of that analysis:
Rebif is modeled as the standard of care (SOC), and it has a total unmet need score of 2.76, as represented by the yellow bar on the left side of the graphic
Gilenya’s unmet need score is 2.50, the yellow bar on the right side of the graphic
Gilenya’s Clinical Innovation, or percent reduction in medical need, is 9.3%: Patients treated with Gilenya have substantially less medical need than patients treated with interferon

The graphic also illustrates Gilenya’s advantages and disadvantages compared with the SOC:
A moderate efficacy advantage (reduction in relapse rates), leading to additional benefits in morbidity
A modest advantage in safety/side effects
A substantial advantage in dosing (oral vs. subcutaneous administration)
Gilenya’s overall Clinical Innovation score of 9.3% is at the top of our “good” range – it was launched in Q4 of 2010, and by the end of 2012 it had achieved annual revenues of nearly $1.2 billion.
When Aubagio (teriflunomide) launched in late 2012, it offered 5% Clinical Innovation relative to Rebif, but it was 5% inferior to the already marketed Gilenya . Aubagio’s sales have lagged.
In March of 2013 Tecfidera (dimethyl fumarate) also received FDA approval in multiple sclerosis. It offers substantialClinical Innovation, of 6.3% above Gilenya (15% over Rebif).

The sources of improvement for Tecfidera over Gilenya are:
A small efficacy improvement
Improved tolerability, and
The improvements in efficacy offer reductions in disease burden
Tecfidera is better than Gilenya, and much better than Rebif. In the fiscal quarter ending June 30, 2013, the Biogen Idec reported sales of $192 million – impressive results for a drug in its first full quarter on the market.